Analysis of Hyper-parameters on Single Cell RNA-Seq Data
scrnaseq
high-dimensional
statistics
regression
Published
May 8, 2024
Code
import numpy as npimport pandas as pdfrom sklearn.decomposition import PCAimport matplotlib.pyplot as pltfrom sklearn.manifold import MDSfrom sklearn.manifold import TSNEfrom sklearn.cluster import KMeansfrom sklearn.mixture import GaussianMixturefrom sklearn.metrics import silhouette_score, silhouette_samplesfrom sklearn.linear_model import LogisticRegression, LogisticRegressionCVfrom sklearn.metrics import silhouette_score, silhouette_samplesimport scanpy as scfrom sklearn.model_selection import train_test_splitimport plotly.graph_objects as goimport plotly.figure_factory as ff
Getting the Data
We will analyze three types of hyper-parameters: Perplexity on t-SNE, number of clusters chosen from an unsupervised method and how these affect the quality of the selected features and how the number of PC’s affect the t-SNE.
To performs these three tasks we are gonna work with real data. Specifically, we’re gonna use scRNA-Seq data from a brain sample (GSM6900730). The data is available on this link.
In our case, let’s do a basic web scrapping code to get the data.
Code
# download the data and load it for the posterior analysisimport urllib.parseimport requestsimport urllibfrom bs4 import BeautifulSoupimport osfrom ftplib import FTPurl ="https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM6900730"response = requests.get(url)soup = BeautifulSoup(response.text, 'html.parser')table = soup.find('table')links = table.find_all('a')download_dir = os.getcwd()for link in links:# Get the URL of the link link_url = link.get('href')# Check if the link contains 'ftp'if link_url and'ftp'in link_url:# Parse the FTP URL link_url = urllib.parse.unquote(link_url) parsed_url = urllib.parse.urlparse(link_url) hostname = parsed_url.hostname path = parsed_url.path# Connect to the FTP server ftp = FTP(hostname) ftp.login() ftp.cwd(os.path.dirname(path))# Extract the file name from the path file_name = os.path.basename(path) local_file_path = os.path.join(download_dir, file_name)withopen(local_file_path, "wb") as local_file: ftp.retrbinary(f"RETR {file_name}", local_file.write) ftp.quit()print(f"Downloaded {file_name}")
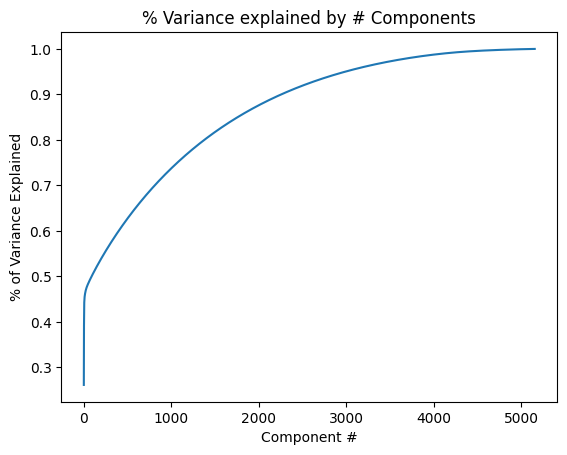
Let’s save the number of components on a variable.
Code
top_pca_components = np.where(csum >=0.85)[0][0]
Analysis of Perplexities
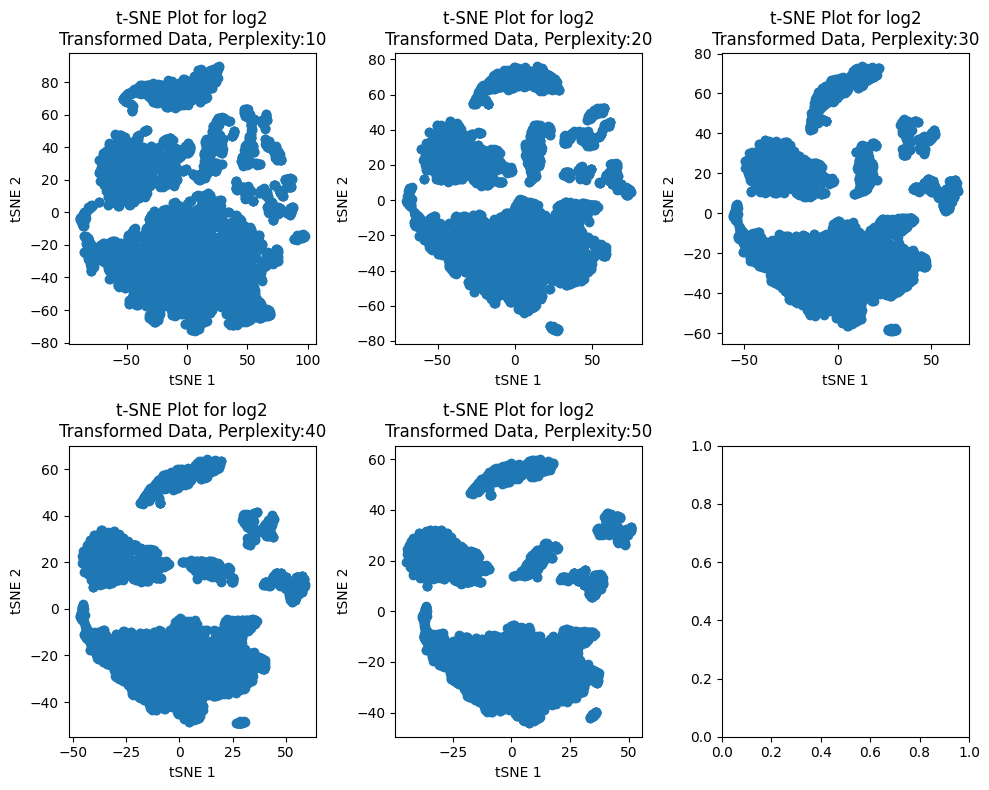
For the first part of the analysis, we are gonna analyze the following perplexities: 10,20,30,40,50 and see how this change the output of the t-SNE analysis.
Code
# let's write a helper function for the tsnedef tsne_plotter(data=None,per=40): tsne = TSNE(n_components=2, perplexity=per) x_tsne = tsne.fit_transform(data) ax.scatter(x_tsne[:, 0], x_tsne[:, 1]) ax.set_xlabel('tSNE 1') ax.set_ylabel('tSNE 2') ax.set_title(f't-SNE Plot for log2\nTransformed Data, Perplexity:{per}')
Code
perplexities = [10,20,30,40,50]num_plots =len(perplexities)num_rows =2num_cols =3fig, axes = plt.subplots(num_rows, num_cols, figsize=(10,8))for i,perplexity inenumerate(perplexities): row = i // num_cols col = i % num_cols ax = axes[row, col] tsne_plotter(pca_axes[:,0:50], per= perplexity)plt.tight_layout()plt.show()
We can see that increasing the perplexity values make the clusters more defined. This is in consonance with the definition of the perplexity parameter which says: “The perplexity is related to the number of nearest neighbors that is used in other manifold learning algorithms. Larger datasets usually require a larger perplexity. Consider selecting a value between 5 and 50. Different values can result in significantly different results. The perplexity must be less than the number of samples”. Also, we can notice that from the perplexity value 30 to 50 there are more defined clusters; specifically, for perplexity 50 we see six more defined clusters so, this is the perplexity value chosen for the following analyses.
Number of Clusters Chosen from an Unsupervised Method
For this part of the analysis on the scRNA-Seq data, we are gonna use the Gaussian Mixture algorithm to get predictec clusters on the data. Then, using these clusters as “labels” we can perform a logistic regression and see how the test score changes when we change the number of clusters.
For this part, we are gonna use the first 2256 principal components with the objective of a precise analysis.
Code
np.where(csum >=0.9)[0][0]
2256
With 2256 PC’s we achieve 90% of explained variability.
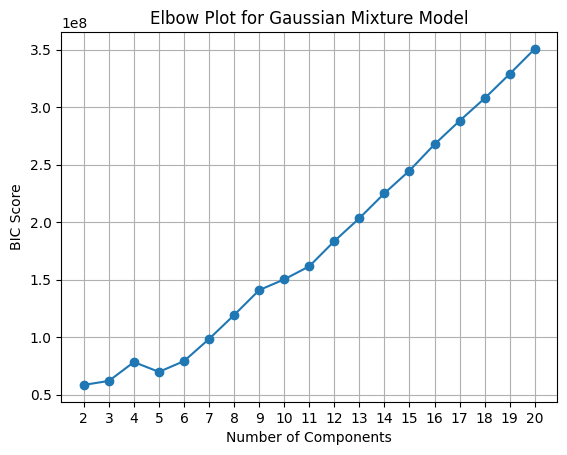
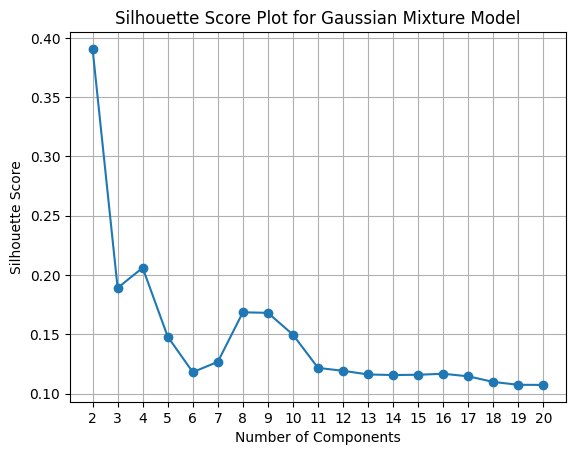
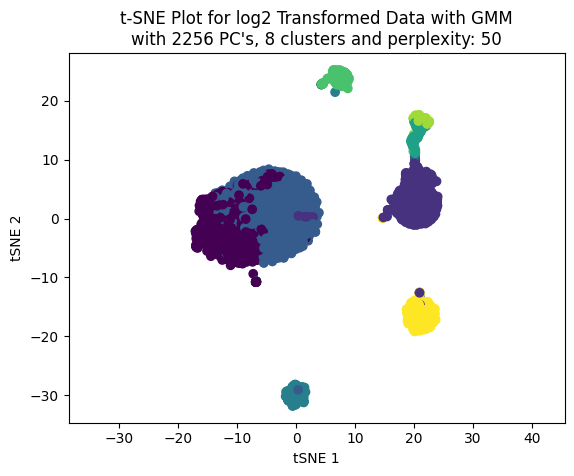
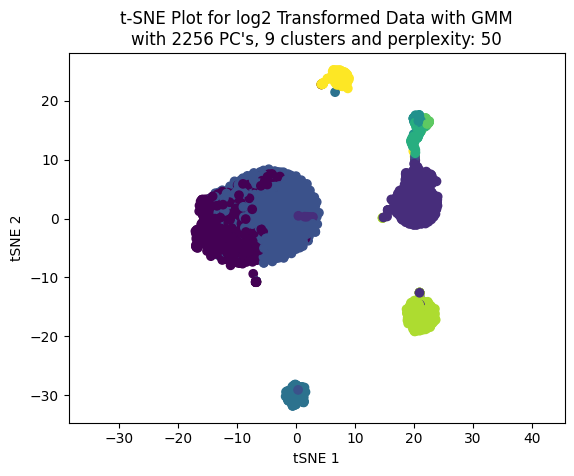
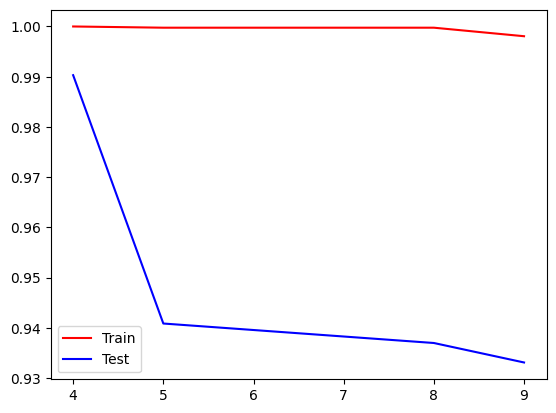
Now, let’s perform the clustering with the GMM model from 2 to 20 clusters. Also, let’s plot the BIC (Bayesian Information Criterion) and the Silhouette plot to see how many clusters we can select.
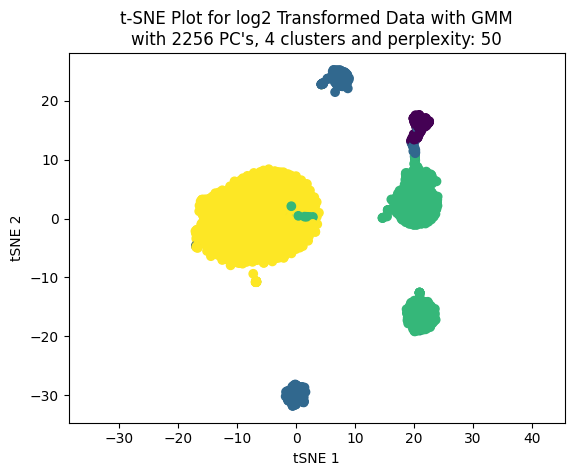
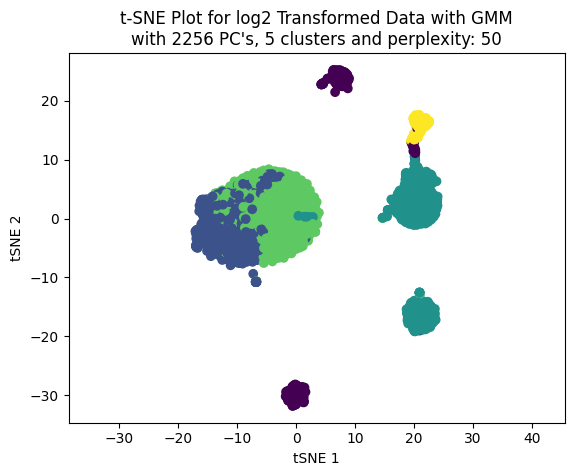
We can see that the best train and test score achieved was using 4 clusters.
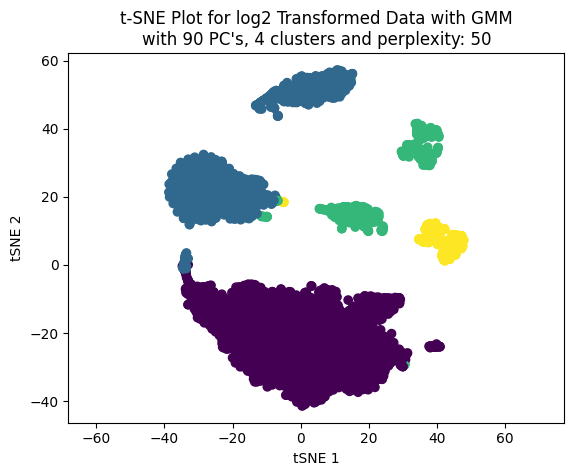
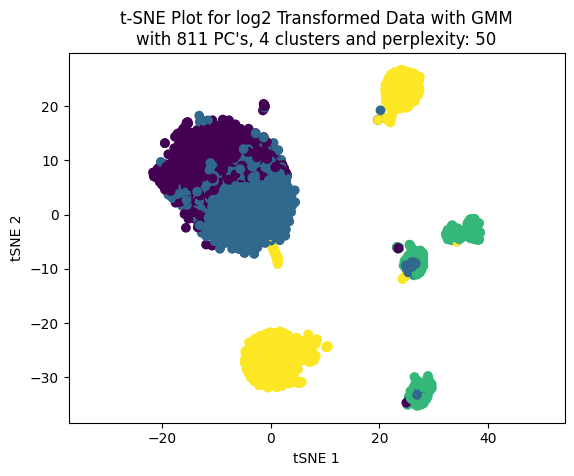
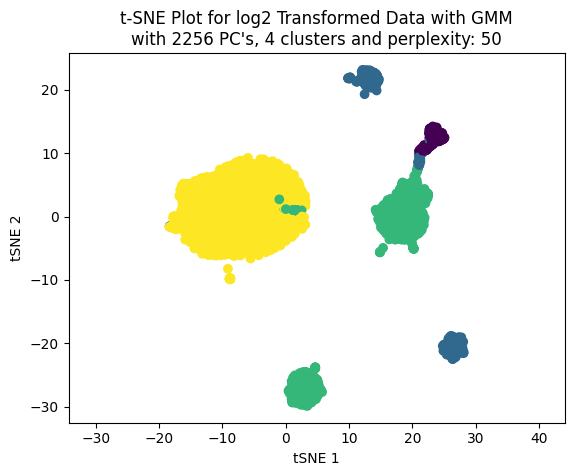
Now, working only with four clusters we can see the effect on the clustering choosing different number of PC’s to build the t-SNE plot.
Effect of PC’s on t-SNE
We cann choose the number of PC’s based on percentage of variance explained. LEt’s work with three values: 50, 70 and 90.
Code
percentages_to_test = [0.5, 0.7, 0.9]pcs_to_test = [np.where(csum >= i)[0][0] for i in percentages_to_test]pcs_to_test
[90, 811, 2256]
Code
np.random.seed(123)for pcas in pcs_to_test: tsne_plotter_pca(n_clusters=4, pca_dim=pcas, perplexity=50)
In practice, the analysis of scRNA-Seq data involves additional steps before and after the dimention reduction. For example, filtering and normalizing, validate the clusters with especific genes (this is because cell types have a specific expression of certain genes and we can identify these cell types with the gene expression related to the cluster) and do downstream analysis like differential gene expression per cell type, gene-set analysis per cell type and more advanced analysis like cell-cell communiation analysis.