This document is part of a series of the analysis of Omics data. Especifically, here is showed how to analyze bulk RNA-Seq data with Bioconductor packages. Also, it’s showcased how to make plots of the RNA data in the context of differentially gene expression and gene-sets.
Keywords
Genomic Ranges, Omics, Bioconductor
1 Introduction
2 GWAS Catalog with a ChIP-Seq Experiment
Here, we are gonna analyze the relation between transcription factor binding (ESRRA binding data) from a ChIP-Seq experiment and the genome-wide associations between DNA variants and phenotypes like diseases. For this task, we are gonna use a the gwascat package distributed by the EMBL (European Molecular Biology Laboratories).
In [1]:
library(tidyverse)
Warning: package 'ggplot2' was built under R version 4.3.2
Warning: package 'tidyr' was built under R version 4.3.2
Warning: package 'readr' was built under R version 4.3.2
Warning: package 'purrr' was built under R version 4.3.2
Warning: package 'dplyr' was built under R version 4.3.2
Warning: package 'stringr' was built under R version 4.3.2
Warning: package 'lubridate' was built under R version 4.3.2
── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 3.5.0 ✔ tibble 3.2.1
✔ lubridate 1.9.3 ✔ tidyr 1.3.1
✔ purrr 1.0.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
library(gwascat)
Warning: package 'gwascat' was built under R version 4.3.1
gwascat loaded. Use makeCurrentGwascat() to extract current image.
from EBI. The data folder of this package has some legacy extracts.
library(GenomeInfoDb)
Warning: package 'GenomeInfoDb' was built under R version 4.3.2
Loading required package: BiocGenerics
Warning: package 'BiocGenerics' was built under R version 4.3.1
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:lubridate':
intersect, setdiff, union
The following objects are masked from 'package:dplyr':
combine, intersect, setdiff, union
The following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabs
The following objects are masked from 'package:base':
anyDuplicated, aperm, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
Position, rank, rbind, Reduce, rownames, sapply, setdiff, sort,
table, tapply, union, unique, unsplit, which.max, which.min
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:lubridate':
second, second<-
The following objects are masked from 'package:dplyr':
first, rename
The following object is masked from 'package:tidyr':
expand
The following object is masked from 'package:utils':
findMatches
The following objects are masked from 'package:base':
expand.grid, I, unname
Loading required package: IRanges
Attaching package: 'IRanges'
The following object is masked from 'package:lubridate':
%within%
The following objects are masked from 'package:dplyr':
collapse, desc, slice
The following object is masked from 'package:purrr':
reduce
The following object is masked from 'package:grDevices':
windows
Warning: package 'AnnotationDbi' was built under R version 4.3.2
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'AnnotationDbi'
The following object is masked from 'package:dplyr':
select
Loading required package: OrganismDbi
Warning: package 'OrganismDbi' was built under R version 4.3.1
Loading required package: GenomicFeatures
Warning: package 'GenomicFeatures' was built under R version 4.3.2
First, we need to download the data, keep the 24 chromosomes (from 1 to Y) and, specify the sequence information from the GRCh38 human genome annotation.
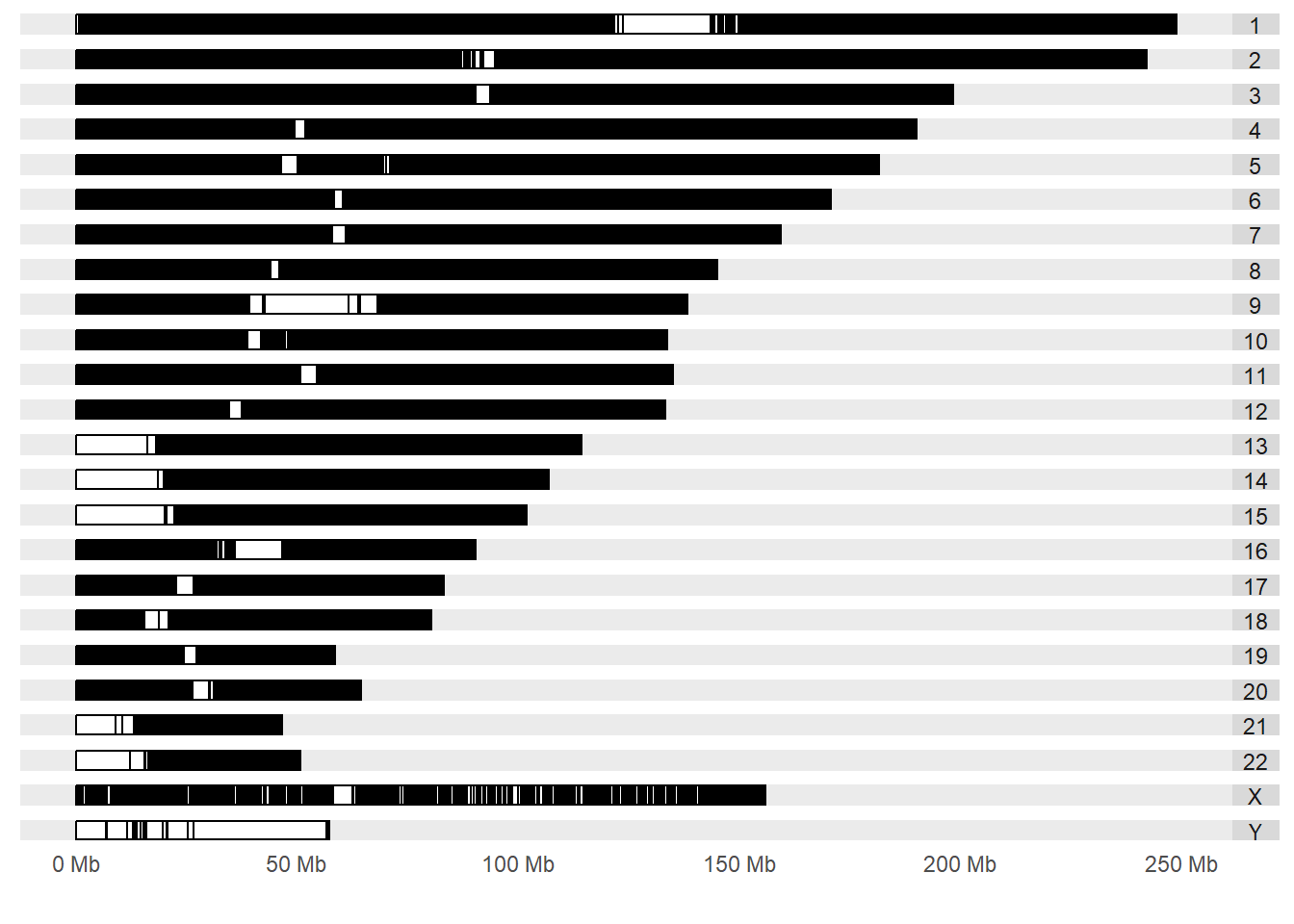
Now, let’s plot a karyogram that will show the SNP’s identified with significant associations with a phenotype. The SNP’s in the GWAS catalog have a stringent criterion of significance and there has been a replication of the finding from a independent population.
In [3]:
ggbio::autoplot(gg, layout="karyogram")
Registered S3 method overwritten by 'GGally':
method from
+.gg ggplot2
Scale for x is already present.
Adding another scale for x, which will replace the existing scale.
Scale for x is already present.
Adding another scale for x, which will replace the existing scale.
If we see the bottom of the GRanges table, this experiment have the hg19 annotation from the human genome. To work on the GRCh38 annotation we need to lift-over with a .chain file. For this we can use the AnnotationHub package.
In [5]:
library(AnnotationHub)
Warning: package 'AnnotationHub' was built under R version 4.3.1
Loading required package: BiocFileCache
Warning: package 'BiocFileCache' was built under R version 4.3.1
Loading required package: dbplyr
Warning: package 'dbplyr' was built under R version 4.3.2
Attaching package: 'dbplyr'
The following objects are masked from 'package:dplyr':
ident, sql
Attaching package: 'AnnotationHub'
The following object is masked from 'package:Biobase':
cache
The following object is masked from 'package:rtracklayer':
hubUrl
We can find overlaps between the GWAS catalog and the ESRRA ChIP-Seq experiment but, there is a problem; the GWAS catalog is a collection of intervals that reports all significant SNPs and there can be duplications of SNPs associated to multiple phenotypes or the same SNP might be found for the same phenotype in different studies.
We can see the duplications with the reduce function from IRanges package:
In [7]:
# duplicated locilength(gg) -length(reduce(gg))
[1] 261160
We can see that there are 261160 duplicated loci. Let’s find the overlap between the reduced catalog and the ChIP-Seq experiment:
We can see 613 hits. Then, we are gonna eobtain the ranges from those hits, retrieve the phenotypes (DISEASE/TRAIT) and show the top 20 most common phenotypes with association to SNPs that lies on the ESRRA binding peaks.
Now, how to do the inference of these phenotype on peaks of these b cells? We can use permutation on the genomic positions to test if the number of phenotypes found is due to chance or not.
In [11]:
library(ph525x)
Loading required package: png
Loading required package: grid
Loading required package: VariantAnnotation
Warning: package 'VariantAnnotation' was built under R version 4.3.2
Loading required package: MatrixGenerics
Warning: package 'MatrixGenerics' was built under R version 4.3.1
Loading required package: matrixStats
Warning: package 'matrixStats' was built under R version 4.3.2
Attaching package: 'matrixStats'
The following objects are masked from 'package:Biobase':
anyMissing, rowMedians
The following object is masked from 'package:dplyr':
count
The following object is masked from 'package:Biobase':
rowMedians
Loading required package: SummarizedExperiment
Warning: package 'SummarizedExperiment' was built under R version 4.3.1
Loading required package: Rsamtools
Warning: package 'Rsamtools' was built under R version 4.3.1
Loading required package: Biostrings
Warning: package 'Biostrings' was built under R version 4.3.2
Loading required package: XVector
Warning: package 'XVector' was built under R version 4.3.1
Attaching package: 'XVector'
The following object is masked from 'package:purrr':
compact
Attaching package: 'Biostrings'
The following object is masked from 'package:grid':
pattern
The following object is masked from 'package:base':
strsplit
Attaching package: 'VariantAnnotation'
The following object is masked from 'package:stringr':
fixed
The following object is masked from 'package:base':
tabulate
library(progress)
Warning: package 'progress' was built under R version 4.3.3
set.seed(123)n_iter =100pb <- progress_bar$new(format ="(:spin) [:bar] :percent [Elapsed time: :elapsedfull || Estimated time remaining: :eta]",total = n_iter,complete ="=", # Completion bar characterincomplete =" ", # Incomplete bar charactercurrent =">", # Current bar characterclear =FALSE, # If TRUE, clears the bar when finishwidth =100) rsc =sapply(1:n_iter, function(x) { pb$tick()length(findOverlaps(reposition(GM12878), reduce(gg))) |>suppressWarnings()})# compute prop with more overlaps than in observed datamean(rsc >length(fo))
[1] 0
3 Explore the TCGA
Please check the dedicated script (top left) to see how to explore and get insights from the TCGA (The Cancer Genome Atlas) database.